Congenital Adrenal Hyperplasia

CONGENITAL ADRENAL HYPERPLASIA (CAH) comes under the medical umbrella of ‘adrenal insufficiency’, along with several other conditions, such as Addison’s disease. In New Zealand, adrenal insufficiency is screened as part of the Guthrie test for all newborns. Congenital Adrenal Hyperplasia is genetic and present at birth (i.e. is ‘congenital’). One in about 10,000 children are born with CAH, though the incidence is higher in some ethnic populations. Although rare it is well understood and good treatment is readily available.
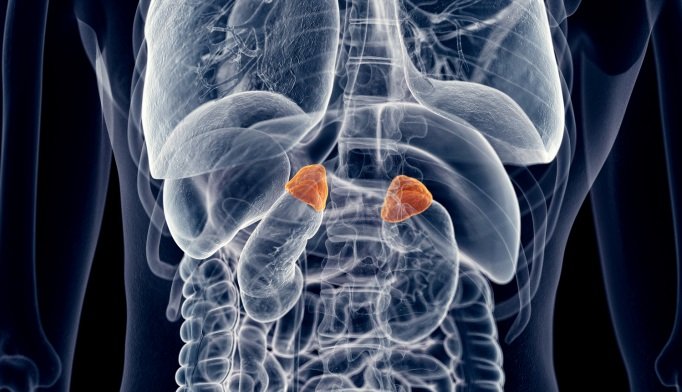
CAH is a deficiency of the adrenal glands, which are situated above the kidneys. Two very important hormones, cortisol and aldosterone, are either partially produced or not produced at all. (All children with CAH lack cortisol but some do produce enough aldosterone.) There are two main forms of the condition, ‘salt-wasting’ or ‘classic’ (SWCAH) and ‘late-onset’ (LOCAH). Early diagnosis provided by the Guthrie test, funded by the New Zealand government, enables families with affected babies to get swift treatment. Late-onset Congenital Adrenal Hyperplasia often appears in early adulthood and is diagnosed by an endocrinologist. Failure to treat this condition can lead to problems with growth and development or even to an adrenal crisis, which may be life-threatening.
How Congenital Adrenal Hyperplasia is treated
CAH is treated with medication taken by mouth, although it can be given by injection if oral medication is not able to be taken. Medication is normally taken two or three times a day in the form of hydrocortisone, prednisone or dexamethasone (to replace cortisol) and fludrocortisone (to replace aldosterone). Due to the body’s delicate hormone balance it is important than the medication is taken in the prescribed doses at the right times.
Children with CAH are under the care of pediatricians and adults receive ongoing care from endocrinologists. Individuals with this condition often enjoy excellent health and are able to lead active, productive lives.